Imodium
Lori Lentowski, BS, MT(ASCP) - St. Joseph’s Medical Center
- Towson, Maryland
Purchase imodium 2mg on-lineAt a constant temperature and stress gastritis nuts safe 2mg imodium, this equilibrium defines the saturated resolution with respect to the designated solid section and respective media gastroenteritis flu generic 2mg imodium amex. Any perturbation in the solute section or solvent part can outcome in a brief metastable state of 139 a hundred and forty either supersaturation (solute > solid) or subsaturation (solute < solid) gastritis diet 600 cheap imodium 2 mg, the place the chemical potentials differ and the system will spontaneously try and gastritis dieta recomendada discount imodium 2 mg without prescription reestablish equilibrium. Any effort to intentionally alter solubility will require a modification in the chemical potentials of both the solute solid state or the solute in resolution. To better understand methods to modify solubility, three key energetic drivers for the solubilization process ought to be considered (2). The second step is the vitality enter essential to overcome solvent�solvent interactions and create a cavity within the solvent which accommodates the solute. The unfavorable power input to this level is then countered with the power launch occurring upon collapse of the solvent cavity across the solute and ensuing intermolecular interactions between solute and solvent. Alterations within the solvent can influence each solvent�solvent interactions and subsequent solvent�solute interactions. This is the premise for many of the cosolvent methods used for solubilization, whereby the solute is decreased shifting the equilibrium toward increased amounts of drug in answer. Solubilization by way of changes within the solid type of a drug (amorphous, polymorphs, and so on. This is probably the preferred strategy for enhancing solubility, but such molecular modifications are difficult to introduce as soon as the drug improvement course of on an entity has been initiated. Hence, molecular design modifications are finest instituted by way of interactions with medicinal chemists within the discovery group previous to drug candidate choice. One of the most generally used methods to present apparent increases in solubility, or complete drug in resolution, is to create different equilibria for the drug or solute to reside in. Creation of alternative equilibria to "sequester" drug offers the premise for solubilization strategies, corresponding to micellar partitioning, chemical ionization, complexation, and partitioning into emulsions. In the simplest of phrases, the solubility of a solute in a given solvent system, as defined by quantity of drug dissolved, appears easily determined, but dependable, reproducible, and significant numbers can be difficult to get hold of. The extra widespread methods are finest described as "fit-for-use," whereby the stable section of curiosity is incubated in solvent and the total quantity of solute current in resolution is measured. The method of solid-phase separation is crucial and really defines the utility of the apparent solubility obtained. Typically, both filtration or centrifugation is used with subsequent assay of filtrate (filtration) or supernatant (centrifugation). Details of separation can be significantly important when colloid scale dispersions exist. Furthermore, as solubilities begin to drop below 1 g/mL, issues of nonspecific adsorption to surfaces (filter, container), coupled with analytical detection limitations, may end up in highly variable values throughout laboratories. Factors corresponding to temperature, power enter, and the character of each the strong phase and the solvent can considerably influence how rapidly equilibrium is obtained. Approaching equilibrium from both a state of supersaturation and subsaturation, taking measurements as a function of time is probably the most effective method. When solubilities are >1 mcg/mL, 24-h incubation will generally approach 90%�95% of equilibrium value, assuming particle sizes are small (3). Implications of Solubility for Parenterals A common problem within the improvement of medicine intended for parenteral administration is the solubilization of a poorly soluble active ingredient (4). For intravenous (intravascular) injection, solubility of the lively ingredient in the plasma must be under saturation upon dilution in order to prevent the potential for precipitation or formation of phlebitis. Injection of a drug into an extravascular website may establish a depot depending on the kind of formulation administered. Drug absorption from a depot by passive diffusion and partitioning depends on drug solubility. A crucial distinction between the pH of the administered drug solution and the physiological pH on the injection site (and/ or solubility of the drug in a cosolvent car and in physiological tissue fluid) may cause an unpredicted lower in absorption as a result of precipitation of the drug on the injection site. Phenytoin is formulated as a sodium salt in a pH 12 resolution of 40% propylene glycol, 10% alcohol and water for injection. When injected into muscle tissue, the large difference in pH and simultaneous dilution of propylene glycol with tissue fluids cause conversion of the sodium salt to less soluble free acid and precipitation at the injection web site. However, amphotericin B is extremely soluble in liposomal intercalation and becomes an integral part of the lipid bilayer membrane. Another commonly studied low solubility drug is paclitaxel with an aqueous solubility of Step 1. The warmth of vaporization at the side of the molar volume of the species, when available on the desired temperature, in all probability affords one of the best means for calculating the solubility parameter. This statement signifies that a solute will dissolve finest in a solvent that has an identical polarity to itself. Strongly polar compounds like sugars or ionic compounds like inorganic salts dissolve only in very polar solvents like water, whereas strongly nonpolar compounds like oils or waxes dissolve only in very nonpolar natural solvents like hexane. The dielectric fixed, solubility parameter, and interfacial/surface rigidity are among the most typical polarity indices used for solvent blending to improve solubility. This reduction is then in comparability with the sphere energy of the charged particle in a vacuum. In basic, polar solvents have greater dielectric constant values than nonpolar molecules. Solvents with a dielectric fixed of lower than 15 are typically thought of nonpolar (7). The dielectric constants of some commonly used solvents and cosolvents in parenteral products are listed in Table 9. Gorman and Hall (10) studied the solubility of methyl salicylate in isopropanol�water mixtures, and obtained a linear relationship between log mole fraction of the methyl salicylate and the dielectric constant of the mixed solvent. For a solution to occur, each solute and solvent molecules must overcome their very own intermolecular attraction forces, so-called van der Waals forces, and find their method between and around one another. This is completed greatest when the attractions between the molecules of both elements are similar. The solubility parameters are defined to categorical the cohesion between like molecules. Hildebrand and Scott include solubility parameters for a number of compounds in their book. A table of solubility parameters has additionally been compiled by Hansen and Beerbower (12), wherein the authors introduced partial solubility parameters D, p, and H. The parameter D accounts for nonpolar effects, p for polar results, and H to categorical the hydrogen bonding nature of the solvent molecules. The sum of the squares of the partial param2 eters gives the entire cohesive vitality density (total) (Equation 9. It is a measure of the work required to create a cavity of unit space of surface from molecules in the bulk, hence relating to cavity formation for solutes. Some surface rigidity and interfacial tension (against water) at 20�C are listed in Table 9.
Purchase 2 mg imodium with visaLeachables can also come up from a reaction of an extractable with drug product or secondary components that will have been ignored when selecting parts for analysis mild gastritis diet cheap imodium 2mg on-line. This is an informational chapter supposed to present a framework for the design gastritis diet 21 imodium 2mg overnight delivery, justification gastritis rash generic imodium 2mg with mastercard, and execution of leachable research for pharmaceutical packaging systems gastritis in pregnancy 2 mg imodium visa. The data and analysis conditions from the extraction research can be used to develop leachable strategies and embody improvement of optimum circumstances for the analytical methods to measure goal potential leachables at required sensitivity. Sample preparation trials can be carried out on the drug product management to optimize the leachable strategies. The technique should be verified for suitability by performing spiking, restoration, repeatability, and linearity research, adopted by validation of the extraction technique and analysis for correct measurements. The leachable strategies ought to be validated based on regulatory tips earlier than routine and stability testing are performed. Several plenty of drug product, saved at completely different orientations, should be evaluated to realize variability and provide adequate information that can be used to (i) determine maximum leachable ranges and establish acceptance standards if necessary, (ii) perform a risk evaluation of leachable species based mostly on precise stability time factors, and (iii) present the power to correlate leachable information to extractables to determine packaging specs, if appropriate. An extraction examine ought to point out greater concentrations of extractables compared to leachables. Management of Extractables and Leachables A correlation could be established if the leachables detected can be quantitatively linked, directly or not directly to an extractable. The most leachable levels can be predicted primarily based on reaching asymptotic ranges of extractables. It is conceivable that routine analysis and control of the packaging components may guarantee acceptable ranges of leachables over the shelf lifetime of the product. In the tip, the container closure system appropriate for one drug product might or is in all probability not appropriate for an additional drug product. Examples are as follows: Dosing cups Sterile empty syringe Calibrated spoons Medicine dropper Since these elements are solely meant for short-term contact (minutes) with drug products, leachables are unlikely; however, whether it is potential and certain that drug merchandise might be saved in these components for a big time (hours or days), corresponding to in a sterile empty syringe or even in a dosing cup, then these parts must be evaluated for leachables. Also, an organic or inorganic liquid utilized in extraction research that may extract chemical components which would possibly be potential leachables but not dissolve the fabric or component being studied. The food simulating solvents are typically water, heptane, and 8% and 50% alcohol. Leachables studies are carried out on the drug product for a time equal to the shelf life. Adhesive from labels on plastic containers have to be thought of as potential leachable however not when on glass containers. Volatile substances from secondary parts can also migrate into primary elements and turn into leachables. The unformulated drug substance which might be subsequently combined or formulated with excipients to produce the drug product. Degradation Product: An impurity ensuing from a chemical change within the drug substance led to throughout manufacture and/or storage of the new drug product by the impact of, for instance, gentle, temperature, pH, water, or by response with an excipient and/or the quick container closure system [4]. Identified Degradation Product: A degradation product for which a structural characterization has been achieved [4]. Impurity Profile: A description of the recognized and unidentified impurities present in a drug product [4]. Identified Impurity: An impurity for which a structural characterization has been achieved [32]. Potential Impurity: An impurity that theoretically can arise throughout manufacture or storage. Process-Related Impurities: Impurities which are derived from the manufacturing process. Aluminum catalysis of epinephrine degradation in lidocaine hcl with epinephrine solutions. The formation of aluminumepinephrine complex and its effect on the addition of bisulfite to epinephrine. Guidance for business - container closure systems for packaging human drugs and biologics ďż˝ chemistry, manufacturing, and controls documentation. Title 21 ďż˝ meals and medicines, chapter 1 ďż˝ food and drug administration, Department of Health and Human Services, subchapter C ďż˝ drugs: basic, part 210 (current good manufacturing follow in manufacturing, processing, packing, or holding of drugs; general) and half 211 (current good manufacturing follow for finished pharmaceuticals). Title 21 ďż˝ food and medicines, chapter 1 ďż˝ meals and drug administration, Department of Health and Human Services, subchapter H ďż˝ medical devices, half 820 (quality system regulation). Title 21 ďż˝ meals and medicines, chapter 1 ďż˝ meals and drug administration, Department of Health and Human Services, subchapter F ďż˝ biologics, half 600 (biological merchandise: general). Title 21 ďż˝ food and drugs, chapter 1 ďż˝ food and drug administration, Department of Health and Human Services, subchapter A ďż˝ basic, part 3 (product jurisdiction), subpart A ďż˝ assignment of agency component for evaluation of premarket purposes, part three. Thresholds and best practices for leachables and extractables in parenteral and ophthalmic drug products working group, work plan. Impact of elastomer extractables in pharmaceutical stoppers and seals; material supplier perspective. Bisharan R, Dicthe D, Feld S-A, Grusgaard J, Patrick D, Seevers B, Smith E, Ulrich D, Weirich W, Womastek K. Guidance for temperature-controlled medicinal merchandise: maintaining the quality of temperature-sensitive medicinal merchandise through the transportation surroundings. Proceedings of the 2nd International Conference on Particle Detection, Metrology and Control, Parenteral Drug Association/Institute of Environmental Sciences. Neutraplex - tubular glass vials with improved hydrolytic resistance for pharmaceuticals. Tygon is a registered trademark of Saint-Gobain Performance Plastics Corporation. Discussion of the physiochemical components that regulate the leaching of organic substances from plastic contact supplies into aqueous pharmaceutical solutions. Modeling of the answer interplay properties of plastic materials utilized in pharmaceutical product container systems. Essential parts of extractables and leachables: from material selection to last report. Polymeric elements and systems used to manufacture pharmaceutical and biopharmaceutical drug merchandise. Characterization of polymeric parts and systems used to manufacture pharmaceutical and biopharmaceutical drug merchandise. Section V Facility Design and Environmental Controls 26 Aseptic Manufacturing Facility Design Mark Caldwell, Robert Helt, Beth Holden, Francesca McBride, and Kevin Schreier Jacobs Engineering Group, Inc. Product kind and presentation influence processing circumstances, tools choice, and due to this fact facility design. The sterile envelope refers to all of the steps carried out throughout and following the final sterile filtration step by way of process completion, which happens after crammed product containers are sealed and a danger of environmental contamination to the product is eliminated. These steps embody ďż˝ ďż˝ ďż˝ ďż˝ ďż˝ ďż˝ Adjuvant, buffer, and media formulation Addition of excipients Adjustment of focus to obtain goal potency Sterile filtration Component preparation Filling, stoppering/plugging, and sealing of product in ultimate dosage containers. When processing organic merchandise, similar to stay virus vaccines, attenuated vaccines, and viral vectors, the biohazard nature of these products place extra demands on the ability.
Buy generic imodium 2mg onlineTherefore chronic gastritis leads to imodium 2mg with visa, it could be more convenient to classify the aggregates when it comes to their size in reference to the capability of varied biophysical and particle analysis technologies gastritis juice fast order 2 mg imodium with amex. Insoluble mixture can additionally be referred to as particle (subvisible and visible-as noted above) and precipitate (large-size species that easily sediments) gastritis young living buy discount imodium 2mg on-line. Finally gastritis chest pain 2 mg imodium amex, discovering the basis reason for aggregation could involve all the above and additional custom-designed protocols. Formation of aggregates may occur under circumstances corresponding to storage, transport, handling, manufacturing, processing, and freezing-thawing. One of the most difficult areas in aggregation, lately, is studying combination formation induced by freezing and thawing of biologicals. Cold denaturation classically refers to denaturation induced by thermal factors per se with no change in the state of the majority and is linked to thermodynamically favored hydration of the hydrophobic core at low temperature [125]. Freeze�thaw-induced aggregation has been linked to secondary factors corresponding to ice surface denaturation, and freeze-induced change in solute concentration and pH. The examine of freeze�thaw-induced protein denaturation and aggregation requires specialised gear and protocols that can probe events in the frozen state. Several aggregate separation strategies are available relying on the type of info sought. Additionally, interference (for larger concentration) and fluorescence (low concentration) probes are also obtainable. Equilibrium studies make use of low centrifugal pressure to obtain a diffusion-controlled equilibrium and are typically used to determine molecular mass in addition to equilibrium binding constants. One of the essential applications of equilibrium research in biotherapeutics is to detect any self-association (reversibly aggregated species). Although the focus restrict could be pushed greater by means of an interference probe, a quantity of sources of "non-ideality" (high focus, sensitivity to excipients, protein form factor) can cripple information interpretation. Several instrument configuration parameters (rotor, cell, loading, probe alignment, wavelength, and so forth. Major functions include separation of drug monomer from larger molecular weight species that may accumulate during storage stability and processing. Also, for a given biologics, the dynamic range for the separation of varied aggregated species is rather restricted leaving giant aggregates unfractionated or misplaced. Prior to initiation of particle migration, the injected sample is focused onto a slim space. For large-size particles (such as protein aggregates bigger than ~60 nm, depending on wavelength of incident laser), angular dependence is critical, and measurement at several angles can produce useful information on measurement. For most protein monomers (<~10 nm), such angular dependence is diminished, and measurement at a 90� angle can be used to determine mass. Larger dimension protein aggregates, with diameters closer to the incident beam wavelength, cause interference (reduction in intensity) of the scattered gentle emanating from completely different elements of the molecules. Mie scattering principles [138] could be utilized for dimension ranges greater than roughly 1/10th of the laser wavelength. Data on molecular measurement can also be obtained within the form of root imply square radius, also referred to as radius of gyration. Angular dependence is diminished when the scale (diameter) of protein monomer or mixture is lower than approximately 10 nm (with incident laser mild wavelength in the visible range), and dimension (diameter) can no longer be decided accurately. With spherical approximation, hydrodynamic radius (Rh) may be extracted from diffusion coefficient values. However, it can resolve species of assorted sizes only if their hydrodynamic sizes differ by greater than two- to fivefold. Dynamic imaging refers to the group of imaging methods that record digital images of particles suspended in fluid. Digital photographs of particles are collected and analyzed to present a digital archive of particle parameters such as Feret diameter, facet ratio, circularity, and depth. Flow-based image recording is very helpful to rely particles in a sure volume and to generate dimension distribution as properly. The optical microscopes used within the dynamic imaging usually have a lower dimension restrict of roughly 1 m. Therefore, dynamic imaging strategies permit complementary size analysis of subvisible in addition to seen particles (up to approximately 300 m or bigger depending on gear model). Although the whole measurement range of the imaging systems is ~1 to >300 m, the person ought to notice that a single measurement might not cover the entire range. When the scale restrict is pushed to cowl the low end, the excessive finish is sacrificed and vice versa. Although numerous flow-based techniques produce comparable data on size/shape of particles, they use totally different methods for sample flow and analysis. Additional variations 238 embrace (i) quality of digital picture, (ii) capability to deal with range of particle concentrations (and need for dilution), and (iii) proportion of particles really analyzed. The recorded photographs of particles may be analyzed to extract a variety of useful physical parameters corresponding to Feret diameter, aspect ratio, circularity, and depth. Building a reference "particle profile" of a protein candidate underneath regular and stress circumstances in numerous formulations can be useful in mitigating future particle occasions. Dynamic imaging offers the good thing about measuring pictures in real time and underneath situations the place particles remain suspended-an benefit over static imaging (such as gentle microscope-discussed later). This might permit superior imaging of extremely irregular shaped particles and monitoring the dynamic conduct of particles if the scale distribution is changing over time. Such info is efficacious during formulation and course of growth of biologics to characterize particles and finding potential preventative measures. Microscopy methods, however, provide direct visualization of morphology along with measurement info. This check includes filtration of an answer using an acceptable membrane filter of 1 m nominal pore measurement to retain the particles, adopted by counting of particles using an acceptable microscope with the beneficial magnification. Particle counting using membrane microscopy, nonetheless, suffers from numerous limitations that include (i) potential detectability points with sure forms of protein particles that are semitransparent, and (ii) particles too fragile to retain their unique measurement upon filtration and subsequent pattern dealing with. Dynamic imaging techniques, as discussed in a earlier part, can provide valuable information on particle morphology in solution situations. This check is predicated on the precept of light blockage and allows for an automatic determination of particle measurement and the variety of particles according to measurement. It is a crucial assay to establish limits of subvisible particles, thereby ensuring high quality and safety of injectable products such as parenteral biotherapeutic formulations [120]. Commercial biotherapeutic merchandise marketed worldwide over the many years have established the good thing about this take a look at to ensure safety [124]. However, emerging research data indicate some limitations of the method as mentioned below. Additionally, new concepts to improve the benefits of this test in addition to updates within the compendial tips are also mentioned in this section. Parenteral formulations with high turbidity or viscosity or particle density might not work on this take a look at. Formulations that are inclined to generate bubbles are prone to produce erroneous (exaggerated) counting.
Purchase 2 mg imodium mastercardEach of them brings a unique resolution to a problem and should gastritis gluten free diet buy generic imodium 2 mg on line be examined in clinic gastritis prevention discount imodium 2 mg fast delivery. However gastritis jaw pain discount 2mg imodium free shipping, it was fully lively when internalized by the host cells gastritis kidney pain order imodium 2 mg overnight delivery, and the linker was cleaved by intracellular proteases that launch the active antibiotic. The use of site-specific conjugations and radiolabeling might provide a extra uniform product with potential benefits within the clinic. As required underneath this approval course of, a randomized section 3 medical study was later carried out in 2004. Certainly, the utilization of engineered amino acids for site-specific conjugation seems to cleared the path and can herald in novel cancer therapies sooner or later. The expiry or impending expiry of patent(s) and data exclusivity intervals for these already-licensed biopharmaceuticals permit potential introduction to the market of lower-cost "copies" of these biologically produced molecules. Government our bodies, tutorial establishments, and medical practitioners have all shown great curiosity in bringing "copies" (called biosimilar products) of biopharmaceuticals expeditiously to market post-loss of exclusivity of the authentic product in order to enhance patient entry to these efficacious-but, very expensive-drugs while concurrently decreasing costs incurred by healthcare methods. The arrival of biosimilars requires engagement between pharmaceutical scientists and healthcare professionals to cascade an understanding of the science underpinning the regulatory approval of a biosimilar product [1]. Elsewhere on the earth, the primary biosimilars have already been accredited, or the regulatory framework wanted to approve biosimilars is in superior levels of enactment. The approval of biosimilars is a extremely regulated process which incorporates extensive similarity assessments (direct comparisons with the originator or reference product) that feed into the "totality of evidence," demonstrating biosimilarity and, finally, in gaining regulatory approval or licensure [2,6,7]. This is a paradigm shift in thinking compared to novel (non-biosimilar) product improvement. As a consequence, the demonstration of analytical similarity with a high diploma of confidence utilizing state-of-the-art methodologies allows reduced nonclinical and medical knowledge packages. The heightened importance of the analytical similarity assessment utilizing sensitive state-of-the-art methodologies to detect minor differences in structure and function in head-to-head comparisons between the proposed biosimilar and its reference product requires that state-of-the-art analytical strategies, expertise, and adequate manpower be obtainable to the developer for the successful conduct of biosimilar product growth packages. Naming of biosimilars and interchangeability/automatic substitution are matters currently beneath dialogue and debate. Biosimilar Development Paradigm Overview In creating a biosimilar for the worldwide market, the first step for a developer is to achieve an intensive understanding of the evolving regulatory necessities in addition to the expectations of national regulatory businesses in areas of the world the place the product is expected to be marketed. Biopharmaceuticals are made by dwelling cells and have intrinsic structural complexities. Additionally, product- or process-related impurities can provoke an immune response [8]. A biosimilar is required to have the same route of administration, dosage kind, and energy as its reference product as specified by the definition of a biosimilar found within the Biologics Price Competition and Innovation Act of 2009 [12], which enabled the biosimilar regulatory pathway within the United States. Glycosylated biosimilars require special consideration to the structure and function of their complex carbohydrate moieties as they are often concerned in the MoA for these medicines corresponding to improve or decrease cell signaling, enzyme exercise, or gene expression. In general, this means that the originator biological product must have been approved for advertising for at least 10 years before a biosimilar could be made out there by another company [3]. The European Commission Consensus Paper "What you want to learn about Biosimilar Medicinal Products" offers a useful supply of further information about biosimilars [15]. Parenteral Medications the proposed biosimilar product to the reference product [6,22]. The expectation for a full Module three reflects the fact that the biosimilar product will probably have distinctive manufacturing processes and control procedures, totally different from that of the reference product. For example, the biosimilar product will likely use different raw supplies, gear, course of controls, and acceptance standards, and probably establish a different formulation and presumably a unique shelf life [6]. Initial Considerations the paradigm shift within the product development process for biosimilar merchandise necessitates consideration of unique requirements. A few of the questions a biosimilar developer needs to address at the outset include ďż˝ Where is the product marketed Originators may not have submitted a license application for a particular product in every nation. From a regulatory perspective, a biosimilar could solely be submitted for approval through the biosimilar pathway if the reference product is approved in that country [7,23]. Understanding the markets in which the reference product is permitted for advertising helps reply the additional questions that observe on this section. While regulators require that a biosimilar has the same strength and dosage type as the reference product, they could not require Drug Substance and Drug Product Process Development Strategy the process development for a biosimilar product follows the same ideas as that of a novel biopharmaceutical. Genotropin is currently marketed in two strengths as auto-injectors, ten strengths as prefilled dual-chamber syringes, and two strengths in vials [27]. In contrast, Omnitrope, an accredited human development hormone biosimilar to Genotropin from Sandoz, is currently marketed in three strengths as auto-injectors, two syringes, and two strengths in vials [28]. Obtaining Health Authority agreement early in development for demonstrating comparability between dosage types and strengths, as well as the number of strengths/ dosage forms to develop for every market, is strongly really helpful. Originators could use multiple manufacturing services to cowl the worldwide distribution of approved/licensed products. An assessment ought to be conducted each as a paper exercise through researching revealed info on originator facility areas and registered production websites for particular products in addition to experimentally through the analytical characterization of the reference product purchased from various regions. Due to the complexity of biologics and the highly specialised processes and equipment wanted to manufacture them, drug substance manufacturing websites are sometimes limited to one to a couple of per product. Typically, the biosimilar pathway requires a biosimilar to be similar to the licensed product sold in that nation. However, a full development program utilizing reference products acquired from a number of markets would make the worldwide development of a biosimilar product extremely challenging from resourcing, budgetary, and logistical perspectives. Many country laws or guidances have enabled an avenue for builders to utilize a single reference product source in every of the stages of the stepwise improvement together with international medical trials by demonstrating similarity of the single selected reference product for international trials to a country/ regionally relevant reference product [3]. Parenteral Medications Sourcing methods for acquiring reference product for both surveillance and medical testing can range from putting orders instantly with the originator company to open market purchases. Sourcing from the open market could result in elevated variety of reference product tons being characterized, which may add to the analytical knowledge set however impose larger burden on human assets in analytical laboratories to perform the additional testing and tracking of samples that might be required. Ideally, the reference product might be blinded during medical research to lower any bias that may materialize from patients or medical practitioners [32]. This is identical challenge that originators face when comparing a new product towards a placebo or a comparator product [32]. In contrast, a typical novel biopharmaceutical product improvement is unlikely to have a comparable established body of data out there to the product sponsor at the start of development. The process begins with the selection of a bunch cell for manufacturing of the proposed biosimilar. Rader outlined over one hundred fifty expression techniques which have been used within the production of biologics in his review of the topic [33]. Biologics and biosimilars manufacturers often utilize platform expression techniques and processes. Typical steps within the developmental pathway for biosimilar product growth by which the reference product lot-to-lot heterogeneity is set by way of purchase of various reference product tons from the open market. Concurrently, extra reference product lots are acquired and characterized with each release type assays and state-of-the-art analytical characterization strategies (discussed additional in the "Analytical Similarity Exercise" section) to discern lot-to-lot variability and heterogeneity of the marketed reference product.
Purchase 2mg imodium with mastercardThe cyclic imide intermediate may be hydrolyzed to yield two potential products: an aspartic acid and an iso-aspartic acid gastritis diet potatoes generic imodium 2mg on-line, which is a beta amino acid gastritis back pain discount imodium 2mg overnight delivery. Both products are acidic variants of the original polypeptide and can be separated utilizing charge-based separation strategies gastritis ibs diet 2mg imodium overnight delivery, and both generate a change in mass of 1 Da relative to the unique polypeptide that can be detected by mass spectrometry gastritis muscle pain discount imodium 2mg with visa. The diploma of degradation relies on many components, including neighboring residues, surface accessibility and conformation, and the pH of the formulation. The susceptibility and fee of deamidation of a given asparagine residue have been shown to be tremendously influenced by the N + 1 residue [6]. Glycine within the N + 1 position has been proven to give the very best fee of deamidation, followed by His, Ser, and Ala. A related mechanism may occur for aspartic acid residues, in which cyclization adopted by hydrolysis to yield both the beginning material or its isomer, the iso-aspartic acid residue, could happen. For these species, the cyclic imide has a net primary shift in cost relative to the aspartic acid starting molecule and is eighteen Da lower in molecular mass. Pyroglutamic acid formation is a standard modification for proteins and happens spontaneously when the N-terminal residue is a glutamine, or less commonly, a glutamic acid. The formation of pyroglutamic acid from an N-terminal glutamine residue generates a net acidic shift and a lack of 17 Da. For monoclonal antibodies, N-terminal glutamine and glutamic acid residues are widespread for each heavy and light-weight chains, and pyroglutamic acid formation is a quite common posttranslational modification for IgG molecules [106]. For monoclonal antibodies, variable ranges of C-terminal lysine on the heavy chains lead to charge heterogeneity as properly. The conserved heavy chain sequence of IgG molecules predicts a C-terminal lysine residue. This residue has been noticed to be removed as a posttranslational modification and is believed to be as a outcome of proteolysis within the cell leading to a heterogeneous population [107]. Typically, a mixture of species exists during which zero, one, or two heavy chains have the lysine removed. Other modifications leading to cost heterogeneity embrace glycation of lysine (acidic shift) [108], carbamylation of lysine (acidic shift), C-terminal amidation (basic shift) [109], and N-terminal acetylation (acidic shift). These potential modifications of biotherapeutic proteins must be detected, quantified, and managed. The analytical methods used ought to be applicable for his or her detection and quantification. There are a wide selection of methods that are helpful for detecting, characterizing, and quantifying charge variants in proteins [110]. However, they give little or no information concerning the varieties or websites of charge heterogeneity present within the molecule. For monitoring stability, inherent charge variability may interfere with the flexibility to monitor degradation using these methods. An instance is the evaluation of deamidation in a glycoprotein in the presence of significant heterogeneity in sialic acid ranges. So, while these strategies could additionally be applicable for routine batch launch and monitoring of consistency, more detailed characterization is required to gain data on the presence of particular modifications resulting in cost heterogeneity. This approach can be used to characterize and quantify, for example, deamidation at a particular website in the presence of inherent heterogeneity elsewhere within the molecule. Proteins will migrate in an electric field to their isoelectric point (pI), which is the pH at which the overall cost is web impartial. Charge variants can be properly separated utilizing this technique, with decision as excessive as zero. For high-resolution separations, a really narrow pH gradient could also be used with lengthy focusing times. This leads to increased resolution as a result of no band broadening happens as a consequence of the mobilization step. Also, detection relies on imaging of the complete capillary, so quantitation is generally extra reproducible. Anion trade resins are positively charged and bind negatively charged analytes, while cation change resins are negatively charged and bind positively charged analytes. In an anion exchange separation, extra acidic, or negatively charged analytes, will be retained extra strongly and can elute later than much less acidic species. Ion change resins may be thought-about sturdy Biophysical and Biochemical Characterization or weak, relying on the kind of resin used. A typical sturdy anion trade resin contains a quaternary amine, which has a fixed optimistic cost or other sturdy basic species, and tightly binds negatively costs species. Weak anion trade resins have primary species such as diethylamine practical groups, which bind negatively charged species, but not as tightly as sturdy anion trade resins. Conversely, cation change resins are both strong acids, corresponding to sulfate groups, or weak acids, similar to carboxymethyl teams. Elution of analytes from ion change resins can be obtained using a salt gradient to compete with the cost on the resin or by altering the charge on the analyte by altering the pH of the mobile section over the course of the separation. This sort of additional characterization can be difficult or unimaginable utilizing gel or capillary electrophoretic strategies. Size exclusion chromatography might have the ability to resolve truncated species as nicely, however this methodology is especially used for the separation of dimers and aggregates from the monomer, discussed beneath. If the truncated species may be resolved appropriately from the primary species, this separation could additionally be an option for explicit situations. Oxidation Oxidation could commonly happen in proteins at exposed methionine residues to form the methionine sulfoxide product. This is considered a serious degradation pathway for so much of proteins and has the potential to impression their structure and performance [11]. In addition, tryptophan and cysteine residues could additionally be oxidized [14,15] and less generally histidine [17]. Some oxidized products may be separated utilizing chromatography, typically reversedphase or hydrophobic interaction. Size Heterogeneity-Truncated Species Size heterogeneity of recombinant proteins might check with truncated variants due to peptide bond hydrolysis or to the formation of aggregates. The formation of truncated species might happen as a consequence of biochemical instability. Formation of highmolecular-mass species is often due physical instability and is mentioned in that part. Physical Stability and Physical Characterization Methods Size Heterogeneity-Aggregates and Particles Aggregation is a course of during which one or more drug molecules combine physically and/or chemically to type non-native oligomers which may stay soluble or turn into insoluble relying on their dimension and different physical properties. Protein aggregates and particles form in a broad range of sizes (nanometer to centimeter, thereby spanning almost million-folds in dimension) and shapes making it extraordinarily difficult to comprehensively characterize particles in a biologics formulation [118�124]. Dimers and different smaller size aggregates are soluble in nature and usually range in dimension from few nanometers to tens of nanometers. The combination species that are in the measurement vary of lots of of nanometers should still stay soluble in the sense that they could not exhibit any change in appearance of the formulation.
Noble Yarrow (Yarrow). Imodium. - Fever, common cold, hayfever, diarrhea, stomach discomfort, bloating, gas, toothache, and other conditions.
- Are there safety concerns?
- Dosing considerations for Yarrow.
- What is Yarrow?
- Are there any interactions with medications?
- How does Yarrow work?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96188
Proven imodium 2mgMany teams are currently evaluating serum-free media choices in order to gastritis loss of appetite purchase imodium 2 mg on line scale back or eliminate this concern gastritis pain after eating generic 2 mg imodium amex. Characterization and Analysis During and after completion of cell manufacture gastritis diet alkaline buy generic imodium 2 mg line, assays should be carried out to show that the product is freed from contaminating organisms and possesses the specified exercise gastritis or gastroenteritis 2mg imodium. Many normal pharmacopeia tests are carried out on the final product along with the assays used to particularly characterize the product. Sterility testing is run on each drug substance and drug product (if these distinct materials are present during manufacture) to fulfill regulators, and multiple variations of the check, relying on geography, could also be run. In addition to testing for the presence of adventitious microbes, mycoplasma testing on the final product should also be carried out. Testing for the presence of adventitious viral contaminants should take place on the final product. In addition, as starting supplies are usually major cells sourced immediately from humans, donor screening must happen. This includes filling out questionnaires and screening for the presence of viruses that can cause infectious diseases. As cells are bodily comparable in size to many particles, attempting to determine and take away particulates in a bed of cells is sort of unimaginable. Visual inspections may be carried out to detect particulates in the completed product before ultimate freezing. Companies should work with manufacturers and suppliers to get hold of materials which would possibly be either low or particulate free. Residuals Residuals are materials that stay in the final product after all manufacturing processes are complete. Some residuals may be faraway from the product by washing the cells prior to last formulation, but some adhere to the cells, which makes it difficult to evaluate their final concentrations. Determining residual ranges can be challenging depending on what the residual material is and the means it interacts with the cells or different materials current as part of the ultimate product. There may not be simple methods obtainable to decide residual ranges, so assay methodology could need to be developed to handle this critical concern. Serum the vast majority of cell therapy merchandise use serum during the manufacturing course of. The concentrations can vary from 1% up to 418 have guidelines for the way tissue must be dealt with and tested in addition to guidance for human cell therapy merchandise. Once a donor sample is qualified, it can then be used for processing to yield the final product. However, as virus might enter via the manufacturing process, adventitious viral screening is carried out on the ultimate drug substance produced (both in vitro and in vivo adventitious screening). Finally, to ensure that the cells manufactured have the desired practical characteristics, viability and potency assays are carried out. As a half of this analysis, cell surface marker screening is commonly carried out to make certain that the cells specific the right markers, which can be indicative of mobile activity as all cells have particular cell surface marker patterns. Based on regulatory steerage, a selected bioactivity assay is required previous to the initiation of Phase three research to show that the cells possess the specified exercise. Depending on their source, further assays might want to be established for evaluating the cells. For example, if cells are thought-about "pluripotent" at some stage throughout their life cycle, then a tumorigenicity assay could also be required. If gene deletions are carried out, then evaluation of off-target results will doubtless be required. Again, as the product itself accommodates particles, this system is troublesome to perform. Another problem to pay consideration to is the presence of different cell varieties that are potentially nontherapeutic. This could be synonymous with a product-related impurity, which is currently dealt with by setting limits for the presence of those cell varieties and potentially testing for their undesirable results (such as tumorigenicity tests for stem cell-derived products). However, right now, no definitive limits are being established because the results and opposed events from varied mixtures might need to be evaluated before appropriate limits may be set. In conclusion, whereas there are particular types of exams which may be commonplace, no matter whether or not the product is a cell therapy or a parenteral drug, there are particular tests that are required to ensure the security and efficacy of the cells being delivered to patients. During the initial freezing process, a managed fee freeze is important to helping protect viability upon thaw and sometimes uses a drop of ~1�C/ min. It is also necessary to add a protein to stabilize the cells during freezing for long-term storage. Stability Stability of conventional biomolecules and small molecules is assessed utilizing non-preferred storage temperatures to take a look at ultimate formulations. Typical non-preferred temperatures would be -80�C, 4�C, 25�C, and 40�C for products frozen at -20�C. Using these storage temperatures would allow the product to be assessed utilizing accelerated stability. If the cells are saved at warmer temperatures, they might begin to become biologically active, and this can slowly cause the cells to deteriorate over time. This fee of degradation depends on temperature and time and must be evaluated as a half of the soundness evaluation for mobile therapeutics. While storage underneath non-preferred circumstances is evaluated to assess stability, real-time stability studies must be performed. Another thing that can be done when working stability research is to use engineering runs to assist stability claims. They must possess high viability and be at a enough concentration to simplify preparation of the frozen product. It is possible to acquire waivers relying on the container dimension, although the precise regulatory body might need to be queried. Devices While cellular therapeutics are often dosed parentally utilizing an intravenous route of administration, using devices for certain indications can lead to the delivery of cells on to the site of harm. If the speed of infusion of cells is important for their impact, syringe pumps may be utilized to aid supply. In the cardiovascular area, units are used to help ship the therapeutic on to the area near the heart. Applications of cells can additionally be carried out after their encapsulation in gels or devices. This protective barrier separates the cells from their environment and forms a "mini-ecosystem" during which the cells can survive for for a lot longer intervals of time. This is especially necessary if an extended duration impact is required, similar to with supply of insulin from pancreatic cells. Shipment of Product Cell remedy merchandise which are transported from the manufacturing facility to a depot, clinical web site, or last dosing web site must stay within the frozen state. Shippers have been developed to perform the shipment sustaining the product at the temperature of -135�C. This saturation allows the dry shippers to have maintain instances of 3�21 days relying on the scale of the shipper (hold times refer to the size of time during which the shipper can retain the desired temperature).
Syndromes - Convulsions
- Vomiting more than once a day
- Time it was swallowed
- Genetic tests
- Injuries
- When does it occur? Evening? Morning?
Discount 2 mg imodium with mastercardStandards or specifications; strategies of testing; and where indicated gastritis symptoms treatment cheap imodium 2 mg without prescription, strategies of cleansing gastritis diet ôĺéńáóę discount imodium 2 mg mastercard, sterilizing gastritis diet ęčíî imodium 2mg without prescription, and processing to remove pyrogenic properties shall be written and followed for drug product container and closures gastritis diet ďůůďäó order imodium 2 mg. The necessities on this part govern the methods used in, and the 542 specified investigational drug, system, or organic product where each are required to achieve the intended use, indication, or impact (another kind of cross-labeled mixture product). Combination merchandise increase a wide selection of regulatory and review challenges, because the merchandise share lots of the similar primary options. When medicine and devices, drugs and biologics, or gadgets and biologics are combined to create a new product, questions are generally raised about how the mix product as a whole shall be regulated. Expectations for patient-focused drug growth, system innovations, and new expedited drug improvement designations have influenced the way packaging and delivery system ought to be qualified to be used. This law was further enhanced supporting patient-centered medical product development in December 2016 when the twenty first century cures act was passed to facilitate faster drug clearances [22]. The kind and amount of container closure info required in each software can differ, and the interpretation of the requirements will depend on various components. Regardless of the product jurisdiction, the underlying precept of threat administration applies. Specifications for packaging supplies and components are present in chapters under 1,000. Parenteral Medications However, these are only a beginning point for qualifying a completed system for supposed use. Batch to batch consistency of packaging elements and acceptance criteria must be primarily based on good scientific principles for each particular system and product [15]. The market package for a drug product contains the first packaging components, secondary package, exterior packaging, and related parts. The concern for bundle componentďż˝product interaction is ranked based on the physical state of the product (liquid vs. Management of Extractables and Leachables ďż˝ Protection: the container closure system shields the product from light, solvent loss, reactive gases, moisture, microbial contamination, and filth. Each suitability category has associated ranges of testing; stage 1 indicates the best diploma of evaluation required. The suitability category for degree 1 security contains extraction/ toxicological evaluation, limits on extractables, and batch to batch monitoring. Extractions could be performed under harsh situations, corresponding to robust solvents at excessive temperatures or gentle conditions, which may or might not replicate the precise circumstances of the final drug product. The packaging guidance offers rationale for a complete examine of high-risk dosage forms such as an injection, inhalation, ophthalmic, or transdermal. It includes two elements: an extraction research on the packaging part to decide which chemical species might migrate into the dosage kind (and at what concentration) and a toxicological analysis of these substances which are extracted to determine the safe stage of publicity through the label specified route of administration. The approach for toxicological evaluation of the safety of extractables ought to be based mostly on good scientific ideas and contemplate the specific container closure system, drug product formulation, dosage kind, route of administration, and dose regimen [15]. A particular section on container closure methods recommends a thorough evaluation of leachables and extractables to consider the capacity of container closure supplies to work together with and modify the therapeutic protein 543 product. The expectation is to conduct a threat assessment to mitigate and management leachables as acceptable. Leachables studies must be performed on the product under stress circumstances as properly as under real-time storage circumstances because in some circumstances the quantity of leachables will increase dramatically over time and at elevated temperatures. Product compatibility testing must be performed to assess the consequences of container closure system materials and all leachables on product high quality. A complete extractables and leachables assessment ought to embody a quantity of analytical methods to assess the attributes of the container closure system that would interact with and degrade protein therapeutic products [25]. Since the packaging steering was issued, the idea of risk-based assessments brought to bear new insights on tips on how to manage extractable and leachable research. It is understood that this steerage is currently being updated to make clear and mirror present day greatest practices. In essence, pharmaceutical growth can be tailored to a prime quality by design (QbD) course of. This begins with defining the quality goal profile, which incorporates the container closure and supply system. Attributes which may be important to product quality must be recognized, justified, and managed. The concept of design area and building quality in through-process growth and enhancements can lead to a level of regulatory flexibility. The important parameters for number of container closure and supply methods must be part of quality danger management course of. Justifications ought to include the following: ďż˝ Suitability of packaging materials primarily based on meant use ďż˝ Integrity of the first container and closure system ďż˝ Protection from moisture, light, and environments, as acceptable ďż˝ Compatibility of the supplies of building with the dosage type (including sorption to container and leaching) 544 ďż˝ Safety of supplies of construction, major and secondary as relevant ďż˝ Suitability of the container closure system for storage and transportation Product and process understanding can employ varied tools corresponding to failure mode results analysis, fault tree evaluation, Preliminary Hazard Assessments, and Hazard Analysis and Critical Control Points [27,28]. Extractable research should be designed to estimate the probability and amount of chemical entities migrating into the drug product to be prioritized for leachable studies. Extractable data can be leveraged to validate leachable methods to decide prevalence and quantity of leaching substances with regard to product high quality and affected person security. Leachable substances must be mitigated and managed as acceptable for a selected drug product. The data collected must be used to establish acceptance criteria, preserve a state of management, and allow continuous enchancment [29]. The improvement and manufacturing means of the drug substance will have the same philosophy as drug product, including the presence of steps designed to reduce impurities. A scientifically justified model can enable a prediction of high quality and can be used to help the extrapolation of working circumstances throughout multiple scales and gear. Adoption of this guideline will promote innovation and continual improvement and strengthen high quality assurance and dependable provide of product, including proactive planning of supply chain changes. Leachables and extractables, though not particularly mentioned, are key in attaining these similar overall quality aims. Another side of danger to product high quality and safety is said to the potential for drug product impurities arising from interrelation with packaging techniques. Impurities are an essential class of crucial quality attributes of completed drug or biologic products. Management of Extractables and Leachables They can embrace organic and inorganic impurities. Leachables are considered impurities in biologics as product-related substances which are molecular variants of the desired product. Process-related impurities can also exist from manufacture gear and include downstream-derived impurities corresponding to column leachables [5,6]. The biocompatibility steerage includes a matrix for choosing applicable checks based mostly on numerous endpoints. These endpoints include cytotoxicity, sensitization, irritation, or intracutaneous reactivity, acute systemic toxicity, material-mediated pyrogenicity, subacute/subchronic toxicity, genotoxicity, implantation, hemocompatibility, continual toxicity, carcinogenicity, reproductive/developmental toxicity, and degradation. Relevant endpoints should contemplate the character of body contact, diploma, frequency, duration (limited/ prolonged/permanent), and circumstances of publicity of the system materials to the body. The degree of toxicological concern must be primarily based on affected person publicity to the chemical entity and the out there toxicological knowledge. One approach is to think about the total patient publicity of the gadget or system element chemical in relation to the quantity at which toxicities are known or in all probability exist. Extraction solvents ought to be chosen to optimize compatibility with the gadget materials and provide info on the forms of chemicals which are more doubtless to be extracted in scientific use.

Imodium 2 mg low costVoltage spikes can happen even if leak paths are clogged with dried product-an benefit not shared with pressurebased analyses that require mass flowing by way of an open leak path to allow leakage detection gastritis diet potatoes purchase imodium 2 mg with mastercard. Therefore chronic gastritis raw vegetables order imodium 2mg free shipping, a quantity of detectors may be stationed at varying heights along a process line to allow for analysis of the complete bundle gastritis peptic ulcers symptoms purchase imodium 2 mg overnight delivery. Laboratory-scale instruments and detectors that allow bundle rotation and whole package scanning are available follicular gastritis definition order 2mg imodium otc, allowing for comprehensive evaluation of an individual take a look at package deal. For instance, the utilized high-voltage present should be excessive enough to allow for enough present to detect leakage paths but not too high that the present arcs over the bundle, on to the bottom electrode, causing false-positive outcomes. Further issues for technique attributes are provided within the Method Validation section of the current chapter. The frequency modulated diode laser output is converted to an amplitude modulation after passing through a gas sample which absorbs at a specific wavelength. The amplitude modulation is proportional to gas concentration and could be phase sensitively detected. The peak-to-peak amplitude of every spectrum is proportional to oxygen concentration. Subsequent measurements of unknown samples use this calibration info to convert measured absorption indicators into significant values of headspace fuel concentration and/or fuel stress. In the case of product sealed with an inert gasoline overlay, leakage of oxygen into the container shall be a perform of diffusive flow driven by the larger oxygen partial stress exterior the container. The results show that holes 5 �m will permit oxygen levels to rise above 1% within 1 day; 2 �m holes will bring about oxygen content larger than 1% after about eight days. Caution is advised, however, when trying to predict package deal integrity for longer durations in accordance with diffusion kinetics. Over time, packages are uncovered to pressure differentials from modifications in altitude or weather and even by doors opening and shutting, all of which drive faster, convective flux leakage, thus complicating such projections. Since the absorption energy of water vapor is 1000X stronger than oxygen in the near infrared, the total space of the absorption profile can be used to determine water vapor focus. In these scans, the entire space is proportional to the moisture partial strain and focus. In principle, any molecule undergoes pressure broadening and can be used for measuring the gasoline pressure in a sealed container. These scans show how the absorption sign broadens as the total gas pressure increases from full vacuum to an intermediate stress and finally to ambiance. Variations in component dimension, elastomer lubrication, gas flushing, stopper insertion, and even handling are only a few of the factors that may affect the outcome. Destructive testing for both oxygen content or vacuum degree utilizing different off-line check strategies is expensive 522 Parenteral Medications the package is placed into cryogenic storage, the gas within the vial shrinks while the external surroundings stays at ambient pressure. This strain differential drives the ingress of dense, cold gaseous setting into the bundle by way of the fashioned breaches. When the vial is taken out of storage, nevertheless, and the elastomer is thawed above its Tg, the material will regain its viscoelastic properties, causing any shaped gaps to reseal and due to this fact trapping chilly gasoline within the vial. As the package deal conditions to ambient, the trapped dense headspace warms and expands, resulting in an over strain within the package deal which could be quantified by laser-based headspace evaluation. Tracer Gas Leak Test Methods-Vacuum Mode Leak detection by tracer fuel analysis is at least as delicate as laser-based headspace analysis. Helium is the most common hint gasoline used for bundle integrity testing, although hydrogen can be used (14, 15). Detection of helium by mass spectrometry is capable of detecting massive leaks of 10 -2 Pa m3/s right down to ultrafine leaks as small as 10 -11 Pa m3/s. Helium trace gasoline testing is most useful for testing leaks in the moderate to ultrafine leak vary. Greatest sensitivity is possible utilizing the vacuum mode, a deterministic take a look at methodology that might be mentioned right here. Tracer gasoline leak checks may be used in the sniffer mode which is probabilistic in nature and mentioned in a future section. In the vacuum mode, a helium-flooded sealed bundle is either totally enclosed in an evacuated check chamber or the seal of the package is isolated inside a take a look at fixture. There are attainable sources of error or technique interferences distinctive to helium mass spectrometry. Steps to prevent elevated helium ranges in the test area embrace proper air flow, distant helium cylinder location, and correct pattern isolation fixturing. Helium also simply permeates via many materials, particularly plastics and some elastomers. Thus, helium permeation by way of the test bundle must be characterized to stop misinterpretation of results. In exams where packages are flushed with helium and then sealed, care must be exercised when massive leaks are suspected as helium could be shortly lost even prior to conducting the take a look at. Finally, sensor efficiency verification utilizing calibrated helium reference leaks is required to ensure accurate outcomes. Research teams led by Kirsch (5, 9, 16) and Nguyen (17) used the helium mass spectrometry vacuum mode to measure the leak charges of optimistic control vials previous to microbiological challenge and vacuum decay leak testing. Evaluation of carbon dioxide headspace content material may point out the presence of microbial growth within a closed pharmaceutical container as microorganisms will expel carbon dioxide as a waste product of cellular respiration. An improve in using ultracold frozen storage or cold chain to keep product potency has prompted analysis to characterize cold temperature impact on package integrity. Even packages well sealed at ambient conditions risk a loss of integrity at conditions under the glass transition point (Tg) of the component supplies, permitting for cold fuel ingress via temporary closure gaps that form. For instance, contemplate a vial that has been capped at ambient situations with atmospheric headspace. All challenge tests require take a look at containers filled with either growth-promoting media or product that helps microbial progress. The product formulation itself or a product placebo is most well-liked as it most carefully simulates the product-package system. However, this may not be sensible if the intention is to validate quite lots of products in similar packaging. The problem vacuum/pressure cycling parameters should at a minimal simulate worst-case pressure variations anticipated throughout product life processing, distribution, and storage. These cycles will improve circulate of packaged media into any leak paths current, thus encouraging potential microbial ingress. For this cause, a valid test requires a comparatively massive population of take a look at samples and constructive controls. Because some expertise is required to design and conduct leak tests by helium mass spectrometry, this expertise is best carried out in a laboratory setting by expert employees. When correctly performed, helium mass spectrometry offers useful information on the quantitative leak price of a package deal. Given the extent of sensitivity, determinations can be made with respect to minute modifications in dimensions or capping parameters on package leakage. Probabilistic Test Technologies Microbial Immersion Challenge Test A microbial immersion problem check process includes filling containers with either growth-supporting media or product adopted by closed container immersion in a bacterial suspension for a predetermined incubation period at circumstances that promote microbial progress. Finally, following incubation, the container contents are inspected for evidence of microbial development either visually or by appropriate analytical evaluation; optimistic challenge organism growth is indicative of bundle leakage. Microbial problem checks through aerosolization, although previously widely employed, are actually thought-about inadequately extreme for the needs of leak detection.
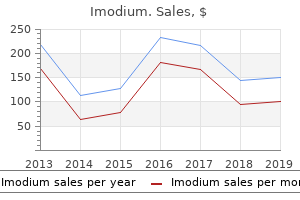
Imodium: 2 mg
Buy imodium 2 mg free shippingEfforts to solubilize medicine are highly depending on both altering the circumstances of the solvent system gastritis help cheap imodium 2 mg line, creating alternative equilibria for the drug to reside in gastritis diet çđĺëűĺ order 2mg imodium visa, altering the macroscopic solid form of the solute gastritis diet treatment imodium 2mg without prescription, or actually changing the solute at the molecular degree gastritis hunger discount imodium 2 mg without prescription. These alterations can enhance the escaping tendency from the stable state, facilitate the cavity formation in the solvent needed for solute insertion, enhance the extent of interactions between the solute and solvent, or simply provide an alternative state by which the molecule can reside. As shall be discussed elsewhere on this book, the final word success of these strategies resides in the capability to deliver the molecule of curiosity to the in vivo milieu with out deleterious outcomes of precipitation upon administration. Thermodynamic investigation of N,N-dimethylformamide/toluene binary mixtures in the temperature vary from 278. Surface rigidity and viscosity measurements of liquids with the survismeter: A single instrumental unit. Determination of the whole and partial cohesion parameters of lipophilic liquids by gas-liquid chromatography and from molecular properties. Solubility of polar natural solutes in nonaqueous techniques: Role of particular interactions. Estimation of the solubility of aliphatic monofunctional compounds in water using a molecular surface space method. Dissolution behavior of crystalline solvated and nonsolvated types of some pharmaceuticals. Supposed isomerism of red and yellow mercuric oxide, and the floor pressure of stable bodies. Comparison of aqueous solubility estimation from aqueous practical group exercise coefficients and the overall solubility equation. Global and native computational models for aqueous solubility prediction of drug-like molecules. Computational mannequin for the prediction of aqueous solubility that features crystal packing, intrinsic solubility, and ionization results. Predictive relationship in the water solubility of salts of a nonsteroidal anti-inflammatory drug. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Initial drug dissolution from amorphous solid dispersions managed by polymer dissolution and drug-polymer interplay. Physical chemistry of supersaturated solutions and implications for oral absorption. Complexation of lomefloxacin with various steel ions and the effect of metallic ion complexation on aqueous solubility. Hydrotropic solubilization of paclitaxel: Analysis of chemical constructions for hydrotropic property. Self-emulsifying drug delivery techniques: Assessment of the effectivity of emulsification. Triglyceride-based microemulsion for intravenous administration of sparingly soluble substances. Amphotericin B in oil-water lecithin-based microemulsions: Formulation and toxicity evaluation. Pertechnetate release from a water/oil microemulsion and an aqueous solution after subcutaneous injection in rabbits. Biotechnol Pharm Asp 2007;6(Solvent Systems and Their Selection in Pharmaceutics and Biopharmaceutics):309ďż˝39. Effect of oil on the level of solubilization of testosterone propionate into nonionic oil-in-water microemulsions. Solubilization effectivity boosting by amphiphilic block co-polymers in microemulsions. Effect of mixed use of nonionic surfactant on formation of oil-in-water microemulsions. Development and characterization of naproxen-chitosan stable techniques with improved drug dissolution properties. Simultaneous impact of cyclodextrin complexation, pH, and hydrophilic polymers on naproxen solubilization. Solubilization of Flavopiridol by pH Control Combined with Cosolvents, Surfactants, or Complexants. Effects of ethanol on formation of inclusion complexes of hydroxypropyl cyclodextrins with testosterone or with methyl orange. Plasma compatibility of injectables: Comparison of intravenous U-74006F, a 21-aminosteroid antioxidant, with dilantin brand of injectable phenytoin. Implanted parenteral drug products are long-acting dosage forms that provide steady launch of the energetic drug substance(s), usually for intervals of months to years. For systemic delivery, they could be placed subcutaneously; for local delivery, they could be placed in a specific region of the physique. Routes of administration for parenteral drug products include intravenous, intraventricular, intra-arterial, intra-articular, subcutaneous, intramuscular, intrathecal, intracisternal, and intraocular. Parenteral dosage forms embody solutions, suspensions, liposomes, emulsions, sterile powders for options and suspensions, implants (including drug eluting), in-situ gels, microparticles and products that include each a drug and a device such as drugeluting stents. Universal tests include: assay, impurities, international and 159 one hundred sixty particulate matter, sterility, bacterial endotoxin, container content material, packaging system, container-closure integrity, and labeling. Specific checks embody: uniformity of dosage type, autos and added substances, antimicrobial preservatives, water content (for dry products), aluminum content, and completeness and clarity of options. Sterile preparations for parenteral use are generally described in accordance with the physical state of the product as follows: 1. Although the term sterile pharmaceutical is applicable to all injections (radiopharmaceuticals included), ophthalmic preparations, and irrigating solutions, this chapter emphasizes the formulation of injectable dosage types. The formulation-the drug itself and the excipients used-must be suitable with body tissues, notably taking care of properties such as hemolysis potential, ache on injection, precipitation of the drug upon administration, and so on. Sterility, lack of pyrogenicity, and absence of particulate matter are different essential concerns from common safety perspective. Various solubilization methods have to be employed to increase the solubility to obtain the required deliverable dose in a minimum possible volume. These techniques embrace use of buffers, salt formation, use of cosolvent, use of surfactants, etc. Stability issues are aimed at growing a formulation that gives sufficient shelf life, which is mostly considered to be the time for 10% degradation. The product is optimized in such a way that its intrinsic degradation pathways, for example, the generally encountered hydrolysis or oxidation, are minimized by appropriate modification of formulation composition, many occasions by utilizing added substances corresponding to buffers and chelating agents. The profitable formulation of an injectable small-volume preparation requires information and expertise to effect rational choices concerning the number of (i) a suitable vehicle (aqueous, nonaqueous, or cosolvent), (ii) added substances (buffers, antioxidants, antimicrobial brokers, buffers, chelating agents, tonicity contributors, and so on. During the course of product development, formulation optimization is an iterative process and evolves because the product strikes from the discovery to clinical to commercial levels. Inherent in the abovementioned choices is the compulsory concern for product safety, effectiveness, stability, and reliability. As the injection formulation is finalized, a selection of extra supportive research must be undertaken to establish ruggedness of the formulation. The majority of parenteral products are aqueous solutions, most well-liked because of their physiological compatibility and versatility with regard to route of administration. However, cosolvents or nonaqueous substances are often required to have an result on solution and/or stability of many compounds.
References - Bretagne S, Marmorat-Khuong A, Kuentz M, et al. Serum Aspergillus galactomannan antigen testing by sandwich ELISA: practical use in neutropenic patients. J Infect. 1997;35(1):7-15.
- Nieuwland R, Berckmans RJ, McGregor S, Boing AN, Romijn FP, Westendorp RG, et al. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95:930-5.
- Konstadt SN, Thys D, Mindich BP, et al: Validation of quantitative intraoperative transesophageal echocardiography, Anesthesiology 65:418-421, 1986.
- Troup GJ: Masers and lasers; molecular amplification and oscillation by stimulated emission, 2nd ed, London, 1963, Methuen. Tucker RD, Ferguson S: Do surgical gloves protect staff during electrosurgical procedures?, Surgery 110:892-895, 1991.
- Puri P, OiDonnell B: Semen analysis of patients who had orchidopexy at or after seven years of age, Lancet 2(8619):1051n1052, 1988.
- Shapiro B: Premature ejaculation: a review of 1130 cases, J Urol 50:6, 1943.
|

